今年12岁的小超(化名),终于可以和同龄孩子一样上学、去享受童年的欢乐,回归正常的生活轨迹。
6年前,年仅6岁的小超开始出现烦躁、间断谵妄,并出现过两次无热惊厥。当地医院诊断为“系统性红斑狼疮”。 但经过规范治疗,病情控制仍不理想,激素减量后症状又复发,再治疗再复发。
辗转2年,小超的病情在协和儿科得到了答案,这是“噬血细胞综合征+肺泡蛋白沉积症”。
协和儿科团队在复盘了小超的病史后发现3个疑点:
1、小超身高发育落后,语言和社会能力发育落后。
2、小超从小不喜欢吃高蛋白的食物,进食后容易腹泻,甚至出现了意识丧失伴高氨血症。
3、小超有着呼吸道反复感染病史。
这些提示着小超可能患有某种遗传代谢病。
实验室检查:尿筛提示尿赖氨酸、乳清酸、尿嘧啶明显升高,基因测序发现了SLC7A7复合杂合突变。
这两项解开了困扰小超多年疾病之迷雾——赖氨酸尿蛋白不耐受症(LPI)。
赖氨酸尿蛋白不耐受症(lysinuric protein intolerance,LPI)又称阳离子氨基酸尿症,是一种罕见的遗传代谢病,呈常染色体隐性遗传。常表现为患儿在摄入含赖氨酸、鸟氨酸等氨基酸的蛋白质后,出现呕吐、腹泻、喂养困难、昏迷、发育停滞、无力等症状。
目前已经报道的 LPI 患者超过 200 名,约 1/3 来自芬兰。该疾病被列入国家卫生健康委员会等5部门联合制定的《第一批罕见病目录》。
病因
赖氨酸尿蛋白不耐受症,病源于SLC7A7基因突变,进而导致赖氨酸、鸟氨酸、精氨酸等氨基酸转运异常,从而发生蛋白质营养不良、血氨增高、肝脾肿大、肺部受累及噬血细胞综合征等免疫性疾病。
辅助检查
B超检查:肝大,脾大。
基因检测:SLC7A7双等位基因出现突变。
血生化检查:空腹时血氨正常,进食高蛋白食物后血氨升高;血赖氨酸、精氨酸、鸟氨酸降低。
尿氨基酸分析:尿液赖氨酸、精氨酸升高。
尿有机酸检测:尿乳酸升高。
口服赖氨酸负荷试验:显示肠道赖氨酸和肾小管吸收障碍。
诊断
由于LPI临床表现不具有特异性,当患者有厌食、呕吐、腹泻、肝脾肿大、生长迟缓、蛋白尿或者肾功能不全等症状时需要鉴别诊断LPI可能性,可通过以下几点进行诊断:
赖氨酸尿蛋白不耐受症家族史。
患儿在断奶、进食富含蛋白质的食物后出现呕吐、腹泻、喂养困难、昏迷等异常表现。
空腹血氨正常,食用高蛋白后血氨升高;血清赖氨酸、精氨酸、鸟氨酸水平降低。
口服赖氨酸负荷试验提示赖氨酸于肠道和肾小管吸收障碍。
基因检测发现SLC7A7基因双等位基因突变。
治疗
LPI治疗目的是通过饮食治疗及药物治疗,以控制高氨血症及并发症,同时保证充足的营养。
饮食治疗
(1)限制天然蛋白质摄入,增加淀粉及脂肪类食物,保证生长发育所需营养供应。
(2)若有血脂异常,可以先尝试饮食调整和补充鱼油。
药物治疗
(1)若发生急性高氨血症危象,可静脉滴注精氨酸、苯甲酸钠、苯丁酸钠等药物,以阻断氨的生成。
(2)待病情稳定后,还需服用祛氨药物。
(3)长期治疗中需补充瓜氨酸、赖氨酸、左卡尼汀等营养素。
遗憾的是本病例中的小超虽然诊断明确,但治疗没有明显进展。截至目前,LPI仍没有成熟的治疗指南,缺少特效药,而小超的不幸远不止于此,重症系统性红斑狼疮还在虎视眈眈她的生命,免疫缺陷导致反复感染,肾功能也持续恶化。
由于LPI传统的饮食疗法收效甚微,协和儿科团队联合南方春富(儿童)血液病研究院,从LPI的发病机制入手,分析可能的治疗方案。在深入了解致病机理后,双方认为虽然国际上尚无对LPI进行造血干细胞移植治疗的文献报道,但从发病机理上来看,异基因造血干细胞移植在治疗上有可行性;从既往经验来看,异基因造血干细胞移植在治疗其他代谢病中获得过成功,可以尝试这一治疗方法,于是创新使用异基因造血干细胞移植治疗LPI。
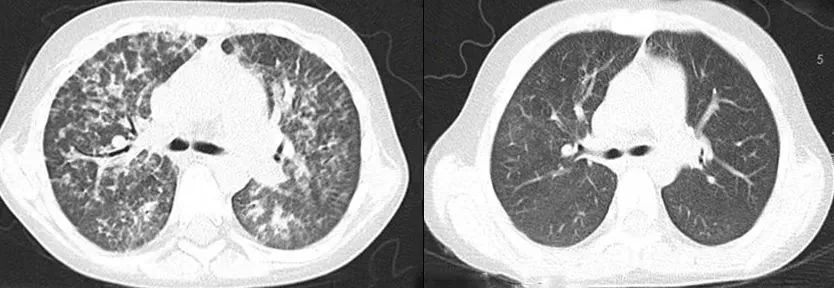
术前(左侧)和术后(右侧)的CT对比
移植后4年中,患儿一直随访治疗,在减停激素及免疫抑制药物后,病情没有复发,没有出现严重的移植相关排斥反应,术前(左侧)和术后(右侧)的CT对比,肺泡蛋白沉积症状显著改善术前(左侧)和术后(右侧)的CT对比,肺泡蛋白沉积症状显著改善,噬血细胞综合征表现迅速缓解,系统性红斑狼疮和肺泡蛋白沉积症的症状显著改善,肝脾明显缩小,代谢、免疫方面各项指标陆续从增高上百倍恢复到正常水平,身体状况平稳。
令人可喜的是,患儿的身高明显增高,逐渐缩小与同龄人的差距。他再次开始了正常的生活。

本病例是国际上首例针对LPI患者进行造血干细胞移植治疗,治疗效果良好。协和儿科团队已将这一研究结果发表在免疫学杂志《过敏及临床免疫学杂志》,为罕见代谢性和免疫系统异常性疾病提供新的治疗思路。


















