罕见病信息网隆重推出《壹周罕见药闻》这一独具特色的品牌栏目。该栏目在每周日更新,以呈现当周罕见病领域的最新消息和下周的罕见病活动预告。我们以更高端、更权威的视角,为您剖析最新、最热门的罕见病信息。
罕药快讯
01 复发性弥漫性间变性星形细胞瘤和胶质母细胞瘤疗法MB101获FDA授予孤儿药资格认定
近日,一款靶向 IL-13Rα2 的 CAR-T 疗法 ——MB-101,获美国食品药品监督管理局(FDA)授予孤儿药资格认定,用于复发性弥漫性间变性星形细胞瘤和胶质母细胞瘤(GBM)的治疗。这款处于Ⅰ 期临床试验阶段的创新疗法,现有数据已展现出良好的耐受性,且 50% 的患者病情达到稳定或更好状态。早期临床中,仅接受 IL-13Rα2 靶向 CAR-T 细胞治疗的 “热肿瘤” 患者出现完全缓解,这一成果为该免疫治疗策略的潜在价值提供了有力佐证。
02 诺华60亿美元王牌罕见病药,III期研究失败!
近日,诺华宣布其IL-17A单抗Cosentyx(司库奇尤单抗)用于巨细胞动脉炎(GCA)成人患者的 III 期研究失败。
GCAptAIN 是一项随机、双盲、安慰剂对照、平行分组、全球多中心 III 期临床研究,在 27 个国家开展,旨在评估司库奇尤单抗治疗 GCA 患者的疗效和安全性。患者被随机分为三个治疗组:司库奇尤单抗 300 毫克、司库奇尤单抗 150 毫克,以及安慰剂组,均联合糖皮质激素 (GC) 减量方案。
03 视网膜色素变性(RP)治疗进阶:MCO-010 向 FDA 启动生物制品许
7月15日, Nanoscope Therapeutics Inc. 宣布,开始向美国食品和药物管理局 (FDA) 滚动提交生物制品许可申请 (BLA),用于其领先的研究性疗法。 MCO-010,用于治疗视网膜色素变性 (RP) 引起的严重视力丧失。
这标志着一个重要的监管里程碑,因为这是第一个为视网膜疾病不区分基因的基因疗法提交的 BLA 。与基因无关意味着 MCO-010 旨在解决 RP 的广泛遗传多样性——该疾病是一种与 100 多个已知基因和 1,000 多个不同突变相关的疾病。
04 家族性高胆固醇血症(HeFH)体内碱基编辑疗法获国家药监局临床试验批准
7月14日,尧唐生物宣布,由尧唐生物与深圳信立泰药业股份有限公司联合申报的YOLT-101注射液临床试验申请(IND,受理号:CXSL2500313)已正式获得国家药品监督管理局药品审评中心(CDE)批准。YOLT-101拟开发适应症杂合子家族性高胆固醇血症(HeFH)。
家族性高胆固醇血症(FH)是由LDL-C代谢的关键基因发生致病性突变所引起一种常染色体遗传病,主要特征为血液中高水平的LDL-C,血管内沉积可导致早发、进行性的动脉粥样硬化性心血管疾病(ASCVD)。FH是最常见的遗传性疾病之一,全球FH患病率约为1/250-1/200,患者总数超3400万;中国FH患者约300万-500万(《中国成人血脂异常防治指南》数据),但诊断率不足5%,治疗缺口显著。
05 5q 型脊髓性肌萎缩症(SMA)基因疗法拟纳入优先审评
7 月 14 日,CDE 官网显示,诺华 Onasemnogene abeparvovec 鞘内注射液(OAV101 注射液,Zolgensma)拟纳入优先审评,这是一种基于腺相关病毒(AAV)载体的基因治疗药物,适用于治疗 6 月龄及以上 5q 型脊髓性肌萎缩症(SMA)患者。
SMA是一种遗传性神经肌肉疾病,由于脊髓前角细胞变性导致肌肉无力和萎缩。2018年5月,SMA被列入国家卫健委等部门联合制定的《第一批罕见病目录》。
06 嗜睡症1型药物3期临床成功
7月14日,武田制药宣布,其同类首创口服食欲素受体2 (OX2R)选择性激动剂药物Oveporexton(TAK-861)在两项全球III期临床试验—FirstLight(TAK-861-3001)和RadiantLight(TAK-861-3002)中成功,在所有剂量下达成全部主要及次要终点,显著改善发作性嗜睡症1型(Narcolepsy Type 1, NT1)患者的多维度症状。这是全球首次在III期研究中确认Orexin(食欲素)受体激动剂的治疗有效性,标志着该机制的临床可行性正式得到验证。
07 国内首个!罕见病眼科基因疗法即将申报上市
近日,Golden Age Health Pte. Ltd. (“GAH”) 与Innostellar Biotherapeutics Co., Ltd. (“朗信生物”) 宣布签署一项为期十年的独家《推广服务协议》。根据协议,GAH将获得朗信生物在中国大陆独家商业化推广其首创新药(BIC)基因疗法候选药物LX101的权利。
LX101是一种腺相关病毒(AAV)基因疗法,旨在将功能性RPE65基因递送至视网膜细胞,以恢复双等位基因RPE65突变遗传性视网膜营养不良(IRD)患者的视觉循环。全球发病率约为每 2 名新生儿 3 至 100,000 人,使其成为新生儿失明的最常见原因之一。在中国,LCA 和视网膜色素变性患者 RPE65 突变的发生率估计为 1% 至 10%,估计患者人数为 8,000 人。
遗传性视网膜营养不良(IRDs)——包括视网膜色素变性(RP)——是一类遗传性疾病,会逐步破坏感光细胞,导致严重的视力损害或失明,通常从儿童期开始发病。RP在全球范围内患病率约为1/3000–1/4000,并被正式列入中国《第一批罕见病目录》(2018年),这充分表明了这类疾病的患者受到了关注。
罕圈动态
01 全球首例!深圳湾实验室联合研发非病毒载体基因疗法治疗DMD进入临床
深圳湾实验室、深圳医学科学院、北京大学深圳研究生院Andrew Lee团队与上海思珀诺因生物科技有限公司的研究人员近日与上海交通大学医学院附属上海儿童医学中心合作,获准开展全球首例采用非病毒载体全长抗肌萎缩蛋白(FL-dystrophin)基因疗法治疗杜氏肌营养不良症(DMD)的临床试验。
这也是国际上首个利用细胞外囊泡递送平台将基因治疗药物导入人体的研究者发起临床试验(IIT)。该项目名为“基于工程化细胞外囊泡(EV)的mRNA新型平台治疗杜氏肌营养不良症”,旨在通过工程化细胞外囊泡递送FL-dystrophin mRNA,评估其对DMD患者的安全性、耐受性及初步疗效,为患者提供更安全、高效的基因治疗选择。
02 中国首个聚焦血友病的患者旅程发布
近日,蔻德罕见病中心发布了关于中国血友病患者疾病旅程的调研相关进展。在罕见病领域,深入了解患者疾病旅程对优化支持政策、改善诊疗现状意义重大。血友病虽在中国发展相对成熟,诊疗体系健全且患者组织经验丰富,但仍需持续探索以提升患者全生命周期的生活质量。
此次调研采用定性研究与定量研究相结合的形式,全面收集全国血友病患者从发病到治疗各个阶段的信息。其价值在于为政府清晰展现患者面临的挑战与困境,助力制定更具针对性的支持政策。通过这一调研,能精准定位患者需求,促进诊疗体系进一步完善,也有助于社会各界更好地理解血友病患者群体。
未来,蔻德罕见病中心计划围绕 “以患者为中心” 的理念持续发力。一方面,将继续深入开展患者疾病旅程调研,扩大疾病种类覆盖范围,同时深度走访患者与相关机构、收集更多实际案例、建立评估体系,推动行业在药物研发、政策制定、患者关怀等多方面,切实从患者需求出发进行优化与创新,携手各方力量,不断改善罕见病患者的生存现状 。
03 资助2000万美元治疗儿童超罕病,诺奖得主押注CRISPR基因编辑 扎克伯格也参与了
近日,陈和扎克伯格基金会(CZI)和创新基因组学研究所(IGI)宣布资助儿科CRIPSR疗愈中心(以下称中心)。中心将联合加州大学伯克利分校的基因编辑专长和邻近洛杉矶分校的临床经验,为患有严重遗传病的婴儿开发治愈性疗法。
新中心将首要重点放在严重免疫出生缺陷(IEI)和严重代谢疾病患儿开发首发个性化CRISPR定制疗法上,第一步先收治8名患者,以实现长期目标:建立“投入使用此种疗法的标准化流程,以便更多家庭得到治疗”, CZI和IGI在通稿中称。
04 总投资60亿元!字节跳动加速布局细胞疗法与高端医院建设
近日,北京爱瑞国际医疗综合体项目在北京市中关村朝阳园北区正式破土动工,该项目标志着美中宜和在被字节跳动收购后,在医疗业务版图中的又一次扩张。
今年年初,北京市卫健委正式批复同意字节跳动在中关村朝阳园北区建设北京爱瑞医院。不久后,北京市发展和改革委员会、北京市住房和城乡建设委员会印发的关于北京市2025年重点工程计划通知中,北京爱瑞国际医疗综合体项目被列为重点工程之一。该项目总投资预计达60亿元,占地约11.37公顷,规划建设总面积为22.4万平方米,规划设置800张床位,预计将于2029年底建成并投入使用。
05 乐城适用于甲基丙二酸血症/丙酸血症(MMA/PA)进口特医食品正式获批
7月14日,在海南省市场监管局、海南省卫健委、海口海关等多部门的通力合作和大力支持下,海南省政府审批同意上海交通大学医学院附属瑞金医院海南医院(海南博鳌研究型医院)临时进口使用唯铎宜特殊医学用途氨基酸代谢障碍配方食品(MMA/PA explore5),标志着国内首款适用于甲基丙二酸血症/丙酸血症(MMA/PA)患者的进口特医食品获批。
该产品的正式获批,是继2025年2月乐城首款临时进口特殊医学用途配方食品Modulen IBD获批后,乐城医疗机构获批的第二款特医食品。截至目前,乐城累计已引进8款特殊食品,其中包括2款特医食品,6款保健食品。
06 北京协和医院开发寡糖糖苷键测序新平台,助力罕见病精准诊断
北京协和医院临床药理研究中心主任韩晓红、主任助理郑昕团队,联合儿科副主任马明圣团队和中国矿业大学(北京)科研团队,开发了基于衍生化和EIEIO技术的寡糖糖苷键测序新平台—“DEED-GL-Seq”,这一技术成功解决了糖链结构解析的核心难题,并在罕见病庞贝病(GSD-II)的精准诊断研究中取得显著成果。相关研究成果于近日发表在国际糖化学与糖组学权威期刊Carbohydrate Polymers上。
07 罕见病诊疗水平提升项目第六期启动 3 家企业中标基因检测服务
2025 年 5 月 28 日,中国医学科学院北京协和医院发布中标公告,中央专项彩票公益金支持的罕见病诊疗水平能力提升项目(第六期)正式启动。该项目自招标公告于 4 月 30 日发布后,经评审专家团队评定,于 5 月 28 日完成定标。
此次项目共有 3 家企业中标,分别是深圳华大医学检验实验室、北京贝瑞和康医学检验实验室有限公司、杭州博圣医学检验实验室有限公司,中标金额分别为 2.415 万元、1.65 万元和 2.31 万元,总中标金额 6.375 万元。
项目服务期为签订合同之日起 1 年,将提供罕见病遗传检测、数据管理、遗传咨询等服务,支持全国协作医院提升诊疗能力。罕见病患者可通过项目协作医院申请基因检测服务,检测样本将通过具备 GPS 跟踪和温控的专业物流运输,确保检测质量符合国家标准。
08 50万美元!罕见病基因组检测计划获资助
7月16日,非营利组织“基因联盟”(Genetic Alliance)周三宣布,其已获得“赫尔姆斯利慈善信托基金”(Helmsley Charitable Trust)一笔50万美元的资助。这笔资金将用于通过该联盟的“iHope基因健康”(iHope Genetic Health)计划,用于提升全球欠发达地区儿童罕见病基因检测可及性,推动其 iHope Genetic Health 计划进一步扩展。
09 砍掉基因疗法,裁员500人!罕见病药企转攻 siRNA,股价反涨30%
7月16日,知名罕见病公司Sarepta Therapeutics宣布了一项重大战略重组计划。该计划包括裁员500人(占员工总数的36%)、调整研发管线重心。这些决定性的变化旨在确保持续的盈利能力,并保持公司履行其为罕见遗传病患者推进创新药物使命的能力。
此次重组是在美国食品药品监督管理局(FDA)要求Sarepta为Elevidys添加急性肝损伤(ALI)和急性肝衰竭(ALF)黑框警告的背景下进行的。ELEVIDYS(delandistrogene moxeparvovec)是第一个也是唯一一个获批用于治疗杜氏肌营养不良症的基因疗法。
10 390亿美元收购而来,罕见病药物未达终点
7月16日,阿斯利康宣布其罕见病药物安塞拉米单抗在AL淀粉样变性的Ⅲ期临床试验中未达到主要终点,股价小幅下跌。心脏淀粉样变性延长生存期(CARES)III期临床项目的结果显示,与安慰剂相比,轻链清除抗体anselamimab在Mayo IIIa和IIIb期轻链(AL)淀粉样变性患者中未达到主要终点的统计学显著性。原发性轻链型淀粉样变已被纳入我国罕见病目录
11 近4000万美元!国内一细胞治疗公司完成B轮融资
7月15日,拓新天成(Drimmunity / Tcelltech)宣布完成近4000万美元B轮融资。本轮融资由国投招商领投,某知名险资和荷塘创投跟投,并获得兴业国信资管、福州闽都人才基金等机构的鼎力支持。澄林资本担任本次融资的独家财务顾问。
本轮募集资金将主要用于支持公司核心管线细胞治疗药物TX-103的中美临床开发,其他管线项目的研发,全球团队拓展,以及加速公司国际化战略布局和推进BD合作。
12 如何让支付方为罕见病疗法买单
近日,Neurvati首席执行官布鲁斯·勒克特强调了将药物开发策略与患者报告的结果相结合的重要性,以在罕见病领域确立价值并确保支付方的支持。勒克特指出,了解和满足患者的特定需求对于证明高价疗法的有效性及其成本合理性至关重要。
《如何让支付方为罕见病疗法买单》(How to get payers to cover rare disease therapies)他强调,罕见病药物的定价和报销必须基于真实世界的患者数据,而不仅仅是传统的临床试验终点。支付方(如保险公司、政府医保)需要看到长期疗效证据和患者生活质量改善的数据,而PROs(患者报告结局)是关键。他建议药企早期与患者倡导组织(patient advocacy groups)合作,以收集更有说服力的数据。
13 遗传病基因检测!两家IVD企业强强联合!
7月10日,宁波海尔施基因科技股份有限公司与厦门百欧迅生物科技有限公司在宁波正式签署战略合作协议。双方将围绕脆性X综合征、脊髓性肌萎缩症(SMA)及其他遗传病基因检测领域展开深度合作,通过技术与资源整合,共同推进精准医疗技术在遗传病防控领域的临床应用,助力实现"健康中国2030"战略目标。
活动预告
01 2025第十四届中国罕见病高峰论坛报名通道开启 | 汉卫罕见 共筑希望
2025第十四届中国罕见病高峰论坛将于2025年9月19日-21日由蔻德罕见病中心联合华中科技大学同济医学院附属同济医院在武汉共同举办,届时将继续以患者为中心,围绕罕见病与孤儿药政策、市场准入及医保支付、罕见病诊疗与研究、商业保险及创新支付探索、孤儿药研发和转化医学、国际合作、患者参与等主题展开深层次研讨。

02 你的研究需要被看见!中国罕见病高峰论坛壁报征集启动
2025第十四届中国罕见病高峰论坛将于2025年9月19日-21日由蔻德罕见病中心联合华中科技大学同济医学院附属同济医院在武汉共同举办。
本届峰会现开启壁报征集,诚邀正在从事罕见病科研的研究机构、医疗机构、患者组织、高校和企业踊跃投稿,积极参与。壁报展览交流活动旨在为我国罕见病医生、科研人员、患者或患者组织、媒体、产业等各相关方搭建一个交流、展示的平台,将集中展示罕见病各领域所取得的最新研究成果、临床进展、罕见病患者生存现状等知识、经验及成果,热忱欢迎踊跃投稿。主办方将从学术性、科普性、创新性等方面给予壁报综合评选。
03 志愿者招募 | 中国罕见病高峰论坛,期待你的力量!
2025第十四届中国罕见病高峰论坛将于2025年9月19日-21日由蔻德罕见病中心联合华中科技大学同济医学院附属同济医院在武汉共同举办,届时将继续以患者为中心,围绕罕见病与孤儿药政策、市场准入及医保支付、罕见病诊疗与研究、商业保险及创新支付探索、孤儿药研发和转化医学、国际合作、患者参与等主题展开深层次研讨。
如果您符合以下条件,可优先入选:
1.有相关公益活动志愿服务经验者;
2.患者亲属、医学相关人员;
3.英文口语优秀者;
4.会摄影,有新闻、医学、英语相关专业背景等。
04 “冀罕守护”为罕见病家庭减免千元基因检测费
全球已知罕见病超7000种,我国患者超2000万。 在河北这片7400万人生活的土地上,预估有数十万罕见病患者在苦苦求医。约80%的罕见病由遗传缺陷导致,基因检测是确诊的“金标准”,但动辄数千元的费用,曾让多少家庭望而却步,延误诊断与治疗?
为打破这一困境,由河北省药学会罕见病专委会联合石家庄金域医学发起的“冀罕守护”罕见病基因检测公益项目,于6月28日在河北省药学会罕见病专委会第八次学术年会上正式启航!
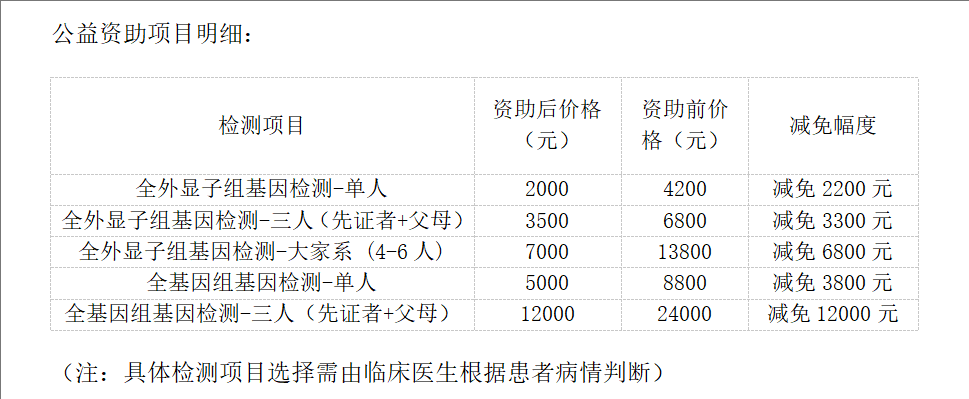
家有疑似或确诊罕见病患者,可通过 “冀罕守护” 公益项目缓解基因检测费用压力,两步即可完成申请:
线上申请:关注 “燕赵罕见病” 官方微信公众号,从菜单栏进入公益项目申请入口,在线填写并提交申请表单。
流程协助:提交申请后,项目组工作人员将主动联系,指导完成材料准备、样本采集等后续流程,助您轻松走完剩余步骤。


















