根据《Translational Neurodegeneration》最新发表的综述,本文将重点探讨反义寡核苷酸(ASO)作为创新药物在神经退行性疾病,特别是在肌萎缩侧索硬化症(ALS)中的发展历程、作用机制及其临床探索。

神经退行性疾病是一类复杂且多因素引发的疾病,涵盖了多种具有独特病理模式、临床表现和潜在致病原因的疾病。这些疾病不仅是导致死亡和残疾的主要原因,还给患者家庭和社会带来了沉重的负担。尽管科学家们已经开发出多种治疗策略,包括蛋白质疗法、基因疗法和干细胞疗法,但目前的治疗方法大多只能缓解症状,尚未能提供明确的治愈方案。基因疗法作为一种有前景的治疗工具,旨在通过修改或调控靶基因的表达来提供神经保护或矫正,已经引起了广泛关注。然而,基因疗法的实际应用仍面临显著挑战,尤其是在中枢神经系统(CNS)中如何实现精准递送和有效转导。
01
在基因治疗中,核酸药物发挥着至关重要的作用,主要包括mRNA药物和小核酸药物。mRNA药物,包括mRNA疫苗,通过进入细胞质翻译特定的蛋白质或抗原,并刺激免疫系统产生免疫反应。而小核酸药物则由短链核苷酸组成,旨在干扰靶基因的表达,从而达到治疗目的。常见的小核酸药物包括反义寡核苷酸(ASO)、小干扰RNA(siRNA)、微小RNA(miRNA)、小激活RNA(saRNA)和RNA适体等。ASO是一类专门设计用于靶向和修改RNA转录本,以减缓或阻止罕见遗传疾病进展的功能性分子。具体来说,ASO是一种人工合成的小尺寸单链核酸,通常长度为13-30个核苷酸。与siRNA相比,ASO在靶向核和细胞质RNA方面具有更大的优势。ASO进入细胞后,根据碱基配对原理与互补的mRNA结合,从而沉默基因的表达。它通过降低靶RNA转录水平,限制有毒蛋白的表达,或者通过剪接调节改变转录序列,恢复正常的蛋白功能。
02
ASO调节基因表达的机制主要有三种方式,具体方式取决于ASO的化学性质和靶位。第一种方式是ASO与mRNA结合后,通过空间位阻的作用,阻止mRNA进入核糖体进行蛋白质翻译,从而下调基因表达。第二种方式是ASO通过碱基配对与靶mRNA结合,招募核糖核酸酶H1(RNase H1)来切割靶mRNA,进而降解其转录产物。第三种方式是通过剪接调节。ASO与前mRNA的外显子结合时,能够通过外显子跳跃或包含,改变剪接模式,这也是最常见的可变剪接类型。例如,与脊髓性肌萎缩症(SMA)相关的SMN2基因因外显子7的跳跃,通常会产生短而不稳定的蛋白质;在杜氏肌营养不良症(DMD)患者中,外显子45和55的缺失会导致产生功能异常的肌营养不良蛋白。用于治疗SMA的外显子插入型ASO药物Spinraza,以及用于治疗DMD的外显子跳跃型药物EXONDYS51和VYONDYS53,均已获得批准。外显子切除后修饰的mRNA可能会导致产生截短的蛋白质,或者通过无义介导的mRNA降解途径降解。当ASO与内含子中的隐蔽剪接位点结合时,可以抑制异常剪接,从而通过正常剪接增加野生型(WT)基因的表达。此外,ASO还可以靶向3'非翻译区(UTR)中的前mRNA的3'poly(A)尾或5'UTR内的帽位点,从而改变蛋白质的合成和翻译。
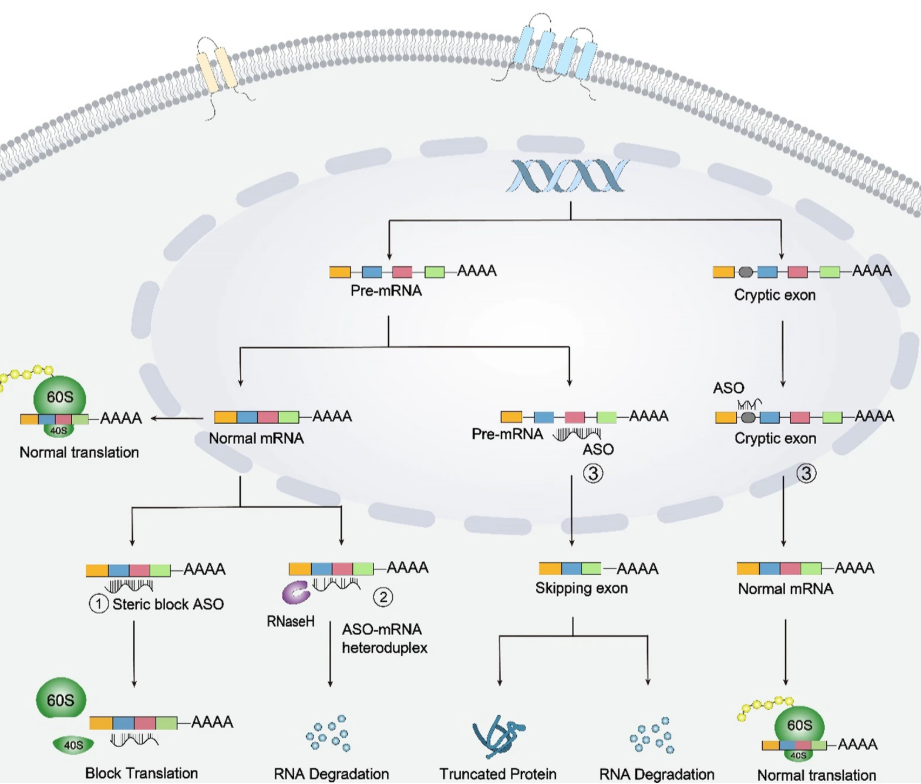
图:ASO药物作用机制
03
肌萎缩侧索硬化症(ALS)是一种进展性神经退行性疾病,主要特征为运动神经元的选择性退化,导致逐渐加重的肌肉无力,最终引发呼吸衰竭。大多数ALS患者在出现症状后的2至5年内不幸去世。目前,约10%的ALS病例为家族性ALS,而其余90%则为散发性ALS。在家族性ALS中,已发现超过30个与疾病相关的基因。最常见的ALS相关基因突变包括超氧化物歧化酶1(SOD1)、TAR DNA结合蛋白(TARDBP)、肉瘤融合基因(FUS)和C9ORF72基因,这些基因的致病变异分别占家族性ALS病例的约60%和散发性ALS病例的约10%。
现有的ALS治疗手段对减缓疾病进展的效果较为有限。此前,FDA已批准四种用于治疗ALS的药物,分别是1995年批准的利鲁唑(Riluzole)、2017年批准的依达拉奉(Edaravone)、2022年批准的Relyvrio以及2023年批准的Tofersen。利鲁唑是一种小分子药物,通过调节电压依赖性Na+通道来减轻运动神经元的兴奋毒性。依达拉奉则是一种小分子抗氧化剂,主要通过减少氧化应激来减缓神经损伤。Relyvrio能够缓解内质网应激和改善线粒体功能(该药物最近已被撤回)。尽管现有疗法有所进展,但疗效仍不尽如人意。为了获得更显著且持久的疗效,分子治疗方法在ALS的研究中日益受到关注,特别是针对ALS相关基因如SOD1、C9ORF72、FUS和ATXN2的毒性功能获得(GOF)的抑制。
针对SOD1基因的ASO治疗
SOD1是1993年首次发现的与ALS相关的基因。SOD1编码超氧化物歧化酶,这种酶在抗氧化防御中发挥着至关重要的作用。然而,SOD1突变导致ALS的具体机制仍不完全明了。研究表明,ALS的毒性可能与突变SOD1蛋白的聚集有关,这暗示着存在GOF机制。
第一个通过RNase H1靶向SOD1 mRNA的ASO治疗是ISIS 333611,这是一种2′-MOE间隙聚体,2007年被FDA授予孤儿药地位。临床前结果促使2010年启动了一项I期双盲安慰剂对照临床试验(NCT01041222),该试验旨在评估鞘内注射ASO 333611在SOD1突变ALS患者中的安全性和耐受性。这项试验是首次将实验性反义药物通过鞘内注射方式用于患者治疗。尽管在脑脊液(CSF)中SOD1蛋白水平未显著下降,但这项试验对于ASO治疗具有里程碑意义,成功展示了鞘内ASO输注在人类中的安全性与潜在有效性,并为ASO从初步选择到临床试验的进一步发展奠定了基础。
BIIB067,也称为qalsody(tofersen),是第二代ASO,针对SOD1前mRNA的不同区域。2020年,I/II期临床试验(VALOR; NCT02623699)显示,受试者接受鞘内注射ASO或安慰剂12周后,未报告严重不良事件,且低剂量和高剂量tofersen组的脑脊液中SOD1蛋白浓度呈剂量依赖性降低。然而,在2016年开始的III期临床试验(NCT02623699)中,经过28周治疗后,患者的临床终点(包括ALSFRS-R、运动功能和肺功能)未显著变化。此后,III期试验转入非随机开放标签延伸试验(NCT03070119),52周时的结果显示,患者的呼吸功能和肌肉强度下降速度有所减缓。基于治疗组患者观察到的血浆神经丝轻链(NfL)水平下降,2023年,tofersen获得FDA加速批准,用于治疗SOD1基因突变引起的ALS患者。
针对C9ORF72基因的ASO治疗
C9ORF72是家族性和散发性ALS中最常见的遗传原因,约占家族性病例的40%和散发性病例的5%。此外,它还是额颞叶痴呆症(FTD)最常见的遗传原因,约占所有病例的25%。为了尽可能减少ASO对C9ORF72正常功能的影响,科学家们研究了针对C9ORF72的ASO——BIIB078(前称IONIS-C9Rx)。2018年,Biogen开始对患有C9ORF72扩增的ALS成人患者开展BIIB078的I期临床试验。NCT03626012是一项随机、安慰剂对照研究,涉及106名患有C9ORF72-ALS的患者(排除了快速进展者),他们通过鞘内注射接受递增剂量的BIIB078或安慰剂。尽管BIIB078的耐受性良好,但该治疗未能达到与ALSFRS-R、慢肺活量和肌肉强度变化相关的任何预定义终点。因此,BIIB078的开发于2022年停止。
另一方面,立体纯寡核苷酸WVE-004在治疗C9ORF72相关ALS或FTD的临床前评估中显示了良好的效果。2021年,一项I/II期临床试验(NCT04931862)启动,旨在评估这种变体选择性、立体纯设计并基于磷酰胍骨架的ASO WVE-004的安全性、耐受性和有效性。然而,在因C9ORF72扩增而患有ALS或FTD的个体中,WVE-004在24周内未表现出临床优势。这一结果促使2023年停止进一步的临床进展。值得注意的是,与安慰剂组相比,WVE-004治疗组脑脊液中poly-GP的水平显著下降了48%,但这一变化并未与功能结果的稳定或改善相关联。
最近,一种名为afinersen的新型化学修饰ASO被设计用于靶向GGGGCC重复扩增周围的内含子区域。该药物通过抑制V1和V3变体的表达,同时保持V2变体在基础水平。这种ASO目前正在进行I期临床试验。
针对FUS基因的ASO治疗
FUS突变约占家族性ALS病例的4%,并与严重且早发的ALS变体相关。FUS基因编码一种RNA结合蛋白,主要参与DNA修复和RNA代谢的调控。针对FUS第六内含子的ASO——ION363(jacifusen)的临床有效性、安全性和药理学特征,目前正在2021年启动的III期临床试验中进行评估。该试验专门针对患有FUS突变的ALS患者(FUSION;NCT04768972)。
jacifusen的III期试验是一项全球多中心研究,涉及64名参与者。在试验的初始阶段,参与者将被随机分配,接受jacifusen或安慰剂治疗,为期29周。随后进入的开放标签期,所有参与者将接受jacifusen治疗,持续73周。预计该研究的最终结果将在2025年公布。
针对ATXN2基因的ASO治疗
转录激活反应DNA结合蛋白43(TDP-43)是由TARDBP基因编码的一种蛋白质,主要在细胞核内表达,发挥着包括前mRNA剪接、mRNA运输及调节mRNA稳定性等多种生理作用。大约97%的ALS病例和45%的FTD病例中,神经元内可见含TDP-43的细胞质内含物。TDP-43蛋白异常的聚集和错误定位在细胞质中,是ALS的关键病理特征。然而,目前尚无针对TDP-43的ASO药物被应用于临床。
ATXN2基因中的三核苷酸重复扩增是ALS的重要危险因素,且通过调节应激颗粒的形成与TDP-43的聚集相关。研究发现,敲低ATXN2基因可以延缓ALS的进展,并显著延长携带TDP-43突变的小鼠的生存期。针对ATXN2的ASO能够逆转核蛋白在细胞质中的错误定位,具有治疗潜力。2020年,一项随机、安慰剂对照的I期临床试验(NCT04494256)启动,评估鞘内注射ASO BIIB105(ION541)的安全性和药代动力学。2022年,该试验进入I/II期,并扩大招募人数至98名参与者,同时增加了包括血浆生物标志物(如神经丝轻链)的疗效终点。然而,尽管BIIB105显著降低了脑脊液中的共济失调蛋白2水平,但并未对血浆神经丝轻链水平以及临床功能指标(如呼吸功能和肌肉力量)产生显著影响。因此,2024年,BIIB105的开发被终止。
04
与小分子药物相比,核酸药物(如ASO)具有更广泛的治疗潜力。它们的设计较为简便,不需要考虑蛋白质的三维结构,从而缩短了开发周期并加速了药物靶标筛选过程。此外,核酸药物通常不会产生耐药性,其疗效通常持续较长时间。随着全球医疗技术的不断进步,像ASO这样的核酸药物在治疗ALS等神经退行性疾病方面展现出巨大潜力。这些药物有望为患者提供更具针对性和有效性的治疗选项,从而显著改善这些疾病的治疗前景。
参考文献:
Ou, K., Jia, Q., Li, D., Li, S., Li, X. J., & Yin, P. (2025). Application of antisense oligonucleotide drugs in amyotrophic lateral sclerosis and Huntington’s disease. Translational Neurodegeneration, 14(1), 4.


















