导语
黏多糖贮积症Ⅱ型 (mucopolysaccharidosis typeⅡ, MPSⅡ) 是MPS中唯一一种X连锁隐性遗传病,又称亨特综合征。MPSⅡ发病率约为1.07/100000,是我国和其他东亚地区最常见的MPS类型,约占所有MPS的一半,已纳入我国《第一批罕见病目录》。

近日,在国际先天性代谢缺陷大会 (ICIEM2025) 上,Regenxbio公司公布其针对MPSⅡ的在研AAV基因疗法RGX-121 (clemidsogene lanparvovec) 在关键性I/II/III期CAMPSIITE临床试验中取得积极结果,为明年的加速审批增添了重要砝码。
|核心数据亮眼,替代终点获得有力支持
在刚刚结束的2025年国际先天性代谢缺陷大会 (ICIEM) 上,Regenxbio公司 (Nasdaq: RGNX) 公布了其用于治疗黏多糖贮积症Ⅱ型 (mucopolysaccharidosis typeⅡ, MPSⅡ, 又称亨特综合征)的在研AAV基因疗法RGX-121 (clemidsogene lanparvovec) 的I/II/III期CAMPSIITE试验的最新积极数据
此前,美国FDA决定将RGX-121的PDUFA审评截止日期,从原定的2025年11月9日推迟至2026年2月8日。Regenxbio已向FDA提交了为期12个月的积极临床数据,以回应RGX-121在BLA审查中提出的要求补充长期数据的要求。
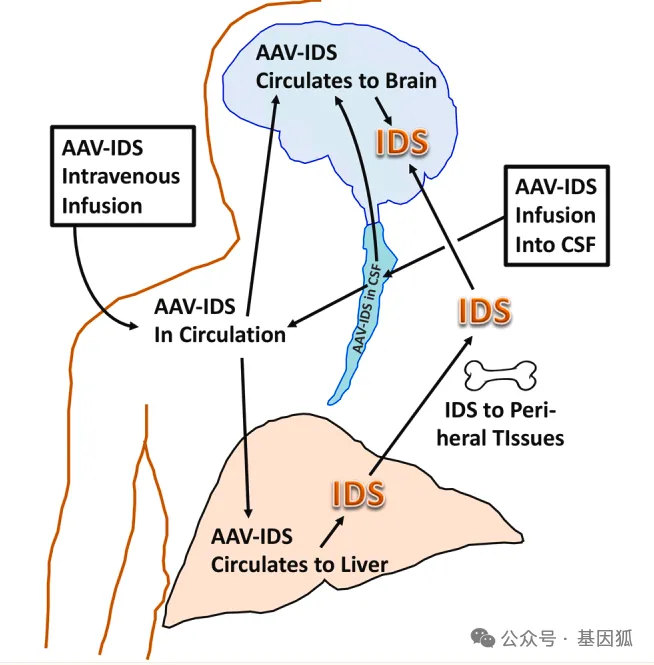
RGX-121 (AAV9.CB7.hIDS) 旨在通过单次鞘内注射,利用AAV9载体将功能性IDS基因递送至中枢神经系统 (CNS),以期从根源上解决MPSⅡ导致的进行性神经功能衰退。此前,RGX-121已先后获得FDA授予的孤儿药、罕见儿科疾病药物、快速通道以及再生医学先进疗法 (RMAT) 等多项资格认定,并获得了欧洲药品管理局 (EMA) 的先进治疗药品 (ATMP) 分类认证。
CAMPSIITE是一项针对4个月至5岁患有神经病变型MPSⅡ的男孩进行的 I/II/III 期多中心、开放标签试验。数据显示,在CAMPSIITE试验的关键队列 (n=13) 中,接受一次性RGX-121治疗的患者,在12个月时脑脊液中关键生物标志物硫酸乙酰肝素D2S6 (HS D2S6) 水平的中位数降幅高达82%,且效果持久。这一数据不仅与此前报告的16周主要终点 (p<0.00010) 数据高度一致,更重要的是,最新分析揭示了16周时HS D2S6水平的降低与1年时的神经认知结局之间存在强相关性。
Regenxbio公司透露,FDA已在2025年8月完成了对RGX-121生物制品许可申请(BLA) 的许可前检查,且无任何观察项 (no observations),这在BLA审查中是一个非常积极的信号。截至目前,FDA未提出任何安全性疑虑,药物在所有26名受试者中均耐受性良好。
值得一提的是,该关键试验采用的是Regenxbio专有的NAVXpress悬浮培养生产工艺平台,可以满足商业化规模的cGMP生产。这表明公司不仅在临床开发上取得重要进展,在生产制造和供应链方面也已为潜在的商业化上市做好了充分准备,解决了基因疗法商业化中常见的产能瓶颈问题。
Regenxbio公司此次公布的12个月长期数据,是AAV基因疗法在中枢神经系统罕见病领域取得的又一重要突破。其核心亮点不仅在于高达82%的生物标志物持续下降,更在于首次在临床上强有力地证实了这一生化指标与神经发育获益之间的相关性。这为后续的监管审批提供了科学依据,也为其他CNS基因疗法的开发提供了宝贵的参考。
目前,神经病变型MPSⅡ尚无有效疗法,存在巨大的未满足临床需求。从临床应用前景来看,RGX-121作为一次性、直达中枢神经系统的基因疗法,具备成为“first-in-class”和“best-in-class”的潜力,良好的安全性和耐受性是其未来被广泛接受的重要前提。如果该疗法最终获批,神经病变型MPSⅡ将从对症支持治疗转向早期、一次性的对因干预,从而最大程度地保留患儿的神经功能,改善其长期生存质量。
黏多糖贮积症Ⅱ型 (mucopolysaccharidosis typeⅡ, MPSⅡ) 是MPS中唯一一种X连锁隐性遗传病,由编码艾杜糖-2-硫酸酯酶 (iduronate 2-sulfatase, IDS) 的IDS基因变异从而导致糖胺聚糖 (glycosaminoglycans, GAG) 在溶酶体中的积累。随着GAG在组织细胞内的逐渐沉积,导致各个脏器功能受损。
虽然MPSⅡ为X连锁隐性遗传病,主要累及男性患 者,但依然有罕见的女性病例被报道。根据患者有无神经系统症状,MPSⅡ型主要分为两种类型: 即有神经系统症状的严重型和无神经系统症状的轻型。
IDS基因位于染色体Xq28,包含9个外显子和 8个内含子,编码550个氨基酸。IDS基因是MPSⅡ的唯一致病基因,但是IDS基因变异的异质性与MPSⅡ患者临床表现的异质性高度相关。
外显子9的p.R468W/Q是我国热点变异常导致重型MPSⅡ,而p.R443X和p.G374G突变常与MPSⅡ轻型相关。值得注意的是,在距离IDS基因着丝粒远端约20kb的位置,存在一个假基因IDSP1,其外显子2、3和内含子2、3、7 的碱基组成和IDS基因高度同源,可产生IDS-IDSP1重排,会给IDS基因变异的诊断带来挑战。
目前,临床上针对MPSⅡ的主要治疗方法包括对症治疗、酶替代治疗 (ERT) 和造血干细胞移植 (HSCT)。而基于ERT和HSCT的MPSⅡ特异性治疗能够显著改善预后,但两者都有一定局限。基因治疗是一种新兴MPSⅡ治疗方法,有望从根本上解决此类问题。
[1]. https://regenxbio.gcs-web.com/
[2]. 陈国庆,张惠文.黏多糖贮积症Ⅱ型的基因诊疗[J].临床儿科杂志,2024,42(3):270-276
版权声明:本公众号署名原创的文章仅为个人学习笔记,且只用于交流学习目的,无意剽窃和抄袭,若涉及版权问题或标记有误,烦请留言联系,将第一时间更正或删除。
免责声明:本公众号对转载、分享的内容、陈述、观点判断保持中立,不对所包含内容的准确性、可靠性或完善性提供任何明示或暗示的保证,仅供读者参考。