摘要
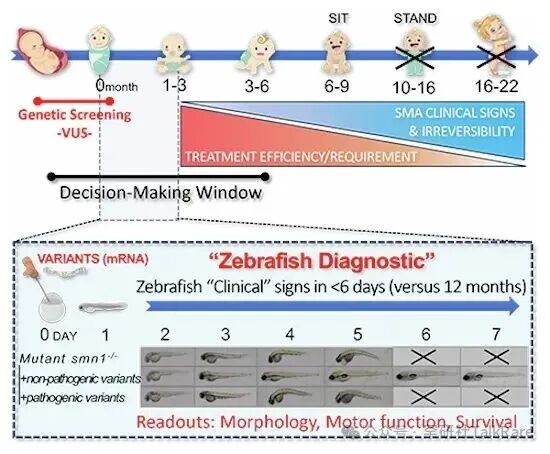
脊髓性肌萎缩症(SMA)由 SMN1 基因功能丧失(LOF)所致,疾病严重程度与体内残存运动神经元生存蛋白(SMN)的水平直接相关。诺西那生钠(Nusinersen)、利司扑兰(risdiplam)及 onasemnogene abeparvovec 基因疗法堪称革命性治疗方案,但此类疗法的最佳应用时机为临床症状出现之前。基于这一特点,产前筛查与新生儿筛查的应用日益广泛,旨在检出 SMN1 基因常见变异位点,明确需要接受治疗的患者群体。
然而,对于新发现的基因变异位点,临床医生缺乏可靠的分析工具,难以在患者出现不可逆损伤前预判其致病性。为填补这一技术空白,本研究构建了 smn1-LOF 斑马鱼模型。该模型会表现出进行性运动功能障碍,且在出生后仅 6 天内死亡。研究人员利用该模型,对两例待确诊新生儿体内检出的 SMN1 基因意义未明变异(VUS)进行致病性评估。
实验结果显示,已知致病性变异位点无法逆转模型鱼的疾病进程;而野生型 SMN1 基因及两例新生儿携带的变异基因,均能有效挽救斑马鱼的脊髓性肌萎缩症核心表型。这一结果证实,该模型在患者诊疗的关键窗口期内,可有效用于 SMN1 基因意义未明变异的致病性检测。
将该检测体系进一步拓展至已知的 SMN1 基因亚效变异(hypomorphs)后发现,此类变异仅能部分挽救模型鱼的疾病表型,其挽救效果显著弱于野生型基因及上述两例意义未明变异,表明该模型亦可区分基因的部分功能丧失效应。
本研究最终判定,这两个意义未明变异位点均为非致病性变异,由此避免了为每名患儿支付超 200 万美元的治疗费用。
除脊髓性肌萎缩症外,本研究为斑马鱼模型作为意义未明变异分析的高效转化医学工具提供了充分的概念验证依据,同时提示该技术手段应纳入临床应用范畴,为疾病诊断与治疗决策提供支撑。