新一代基因编辑技术正从逐个修复突变的思路,转向通用型疗法,有望开发出成本更低、应用范围更广的药物。

图:许多罕见病存在共同的基因缺陷,生物技术公司正利用这一特点,开发新型基因编辑工具。
首批以不依赖特定突变类型的方式修复基因缺陷的基因编辑工具,即将进入临床应用阶段。由 Flagship Pioneering 投资的两家公司 ——Tessera Therapeutics 和 Alltrna,正准备在人体中开展新型基因药物试验。
Tessera 刚刚宣布与再生元(Regeneron)达成 1.5 亿美元合作,随后便获得了美国和澳大利亚监管机构的批准,启动其基因写入疗法的临床试验。

与此同时,Alltrna 将于 2026 年在美国提交研究性新药申请(IND),其产品为工程化转运 RNA(tRNA),可纠正共有的提前终止密码子,进而恢复蛋白质合成,且不受具体致病蛋白种类限制。

这些研究方案标志着一个可喜的转变:罕见病治疗正从个体化 “单一患者(N-of-1)疗法”中走出来。
KJ 婴儿的案例令人瞩目:他在确诊仅 6 个月后,就通过治疗纠正了尿素循环基因上的特定突变。然而,尽管这类个体化疗法效果非凡、改变人生,但对患有遗传性罕见病的患儿家庭而言,却难以带来普遍安慰 —— 因为有能力开展这类昂贵个性化疗法的中心寥寥无几。
要让罕见病治疗不再罕见,需要技术专长、监管改革与资金三者结合。而这样的协同局面,或许已近在眼前。
基因编辑正在进入“不依赖疾病类型” 的时代。
生物技术公司如今开发的基因编辑药物,不再只针对某一种疾病,而是能跨多种疾病起效。正如博德研究所、哈佛大学与霍华德・休斯医学研究所的刘如谦(David Liu)所述:

“整体策略是开发一些新方法,让基于基因组的治疗手段不只适用于携带某一种特定突变的患者。”
已知有超过 7000 种疾病由基因错误导致,若为每一种都开发专属疗法,将是极其艰巨的挑战。
不依赖特定突变的疗法可通过解决基因功能中共通的问题来简化这一难题。
通过发现并攻克疾病背后的共性,不依赖疾病型疗法(disease-agnostic therapies)能用同一种药物惠及大量患者 —— 即便他们在同一基因上携带不同突变。
这类通用策略中最前沿的方向围绕转运 RNA(tRNA)展开
多家公司正利用一类 tRNA 治疗因突变产生提前终止密码子的疾病:这类突变会中断翻译,导致必需蛋白无法合成。
经过编辑或工程化改造的抑制型 tRNA(suppressor tRNA),能将 “终止密码子” 识别为 “有义密码子”,从而让完整蛋白得以合成。
鉴于由错误终止密码子引起的疾病占所有遗传病的 11%,能挽救蛋白翻译的抑制型 tRNA 长期以来都是药物研发的重点目标。
tRNA 是小型、结构高度稳定的分子。作为治疗手段,它们可在体外工程化后给药,恢复缺失的蛋白合成;也可在体内编辑,实现抑制型 tRNA 的永久表达。
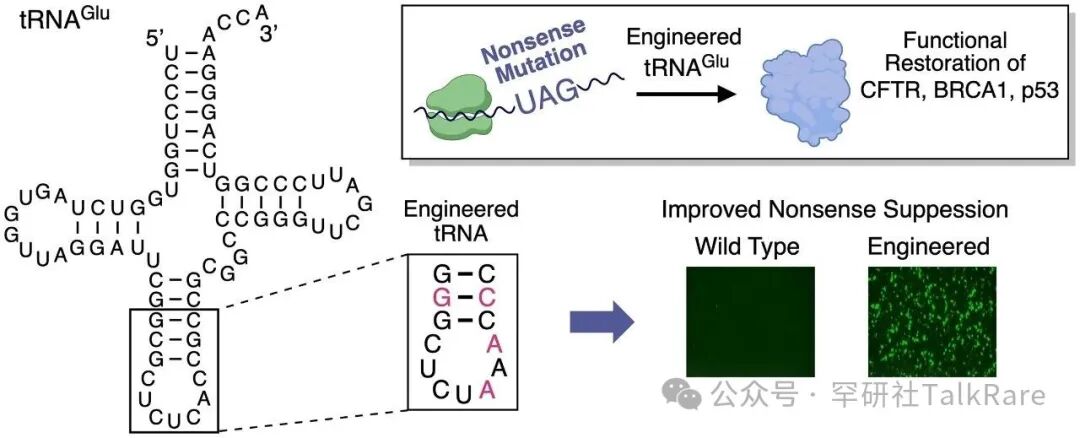
图:一种用于有效抑制致病性无义突变的工程化谷氨酸tRNA(引自:PMID: 40539513)
Alltrna 首席执行官 Michelle Werner 表示,体外方案的优势在于不造成永久性基因改变;剂量明确、可调节、可按需重复给药,若出现安全性问题甚至可完全停止。
与之相对,开发了体内方案的刘如谦则指出,创建永久性抑制型 tRNA 的优势是一次性治疗—— 鉴于 tRNA 半衰期很短,可避免终身、频繁给药。
Alltrna 有望成为首家将工程化抑制型 tRNA 用于人体的公司。
其 tRNA 设计包含三个独立层面:反密码子:实现对特定提前终止密码子的通读;反密码子外侧序列:提升翻译效率与稳定性;化学修饰。
其首个候选 tRNA 药物 AP003,用于治疗一种未公开的肝脏相关疾病,这类疾病包括由提前终止密码子突变引起的苯丙酮尿症、甲基丙二酸血症等。Alltrna 通过肝脏靶向脂质纳米颗粒递送治疗性 tRNA。
为靶向肝脏以外的组织(如肌肉、心脏,最终扩展至大脑),Tevard Biosciences 选择了腺相关病毒(AAV)递送。
该公司由 Daniel Fischer 与 Warren Lammert 创立,二人均有子女患有Dravet 综合征(一种罕见癫痫,很大一部分病例是由提前终止突变导致)——Tevard 正是 Dravet 的倒写。
公司首席科学官(CSO) Elisabeth Gardiner 表示,他们通过筛选近 8 万种新型抑制型 tRNA,并整合多种天然修饰以优化通读效率,鉴定出一组抑制型 tRNA:在杜氏肌营养不良小鼠模型中体内恢复全长抗肌萎缩蛋白,在由诱导多能干细胞分化的人心肌细胞、以及由 TTN 截短导致的扩张型心肌病小鼠模型中,恢复全长肌联蛋白。肌联蛋白是肌肉骨架的一部分,也是已知最大的蛋白,其缺陷影响庞大患者群体。
Gardiner 说:“我们将能在更多患者中开展测试,并理解抑制型 tRNA 如何不仅在儿童患者、也在成人患者中发挥作用。”
刘如谦团队并未递送 tRNA,而是使用先导编辑(prime editing)—— 一种基于 CRISPR、可精准实现靶向 DNA 改变的基因组编辑技术 —— 将抑制型 tRNA 基因永久整合进基因组。

这项技术名为 PERT(先导编辑介导的提前终止密码子通读),可将无义突变转变为通读密码子,优化 tRNA 生成,从而产生足够蛋白以挽救病理表型。
PERT 已在 6 种携带不同终止密码子的人类体外细胞模型中起效。
“我们治愈了 GFP 缺陷小鼠,” 他打趣道 —— 指的是他们用插入绿色荧光蛋白转基因的小鼠模型测试编辑平台。
该工具还单次治疗就挽救了黏多糖贮积症Ⅰ型(亦称为 Hurler’s syndrome)小鼠模型。Hurler’s syndrome 是一种溶酶体贮积症,由 IDUA 基因突变导致酶功能丧失,若不治疗通常 10 岁前死亡。
团队证实,新 tRNA 重启了蛋白合成,却未干扰正常蛋白翻译—— 这是外源 tRNA 大量涌入时令人担忧的问题。
刘如谦还指出,由于 PERT 将可替代、冗余的内源性 tRNA 转变为新型抑制型 tRNA,它保留了 tRNA 基因天然的基因组调控环境,有利于提升 tRNA 效力。
将基因大小的 DNA 片段插入人类基因组依然困难重重,但一旦实现将具备巨大优势
刘如谦说:“靶向基因整合原则上可帮助同一疾病、但不同突变的患者 —— 有些疾病存在数千种不同突变,均导致某一蛋白功能丧失。”
他的团队改造了数十年前发现的可移动遗传元件,开发出两种策略:1. 先用先导编辑插入重组酶着陆位点,再用实验室进化的重组酶将目标基因插入该位置;2. 使用 CRISPR 相关转座酶 —— 通过噬菌体辅助连续进化(PACE)系统构建 —— 将目标基因插入基因组。
后一系统与哥伦比亚大学 Sam Sternberg 合作开发,在多种人类细胞类型中实现高达 30% 效率的靶向整合。两种系统均可将大于 10,000 bp的基因大小 DNA 片段精准插入人类基因组的靶向位置。
Patrick Hsu 是 Arc 研究所创始人,同时任职于加州大学伯克利分校生物工程系。他从自然界中寻找可插入大片段序列的元件,并锁定了大型丝氨酸重组酶。
这类酶的功能专一:以固定方向将供体 DNA “扣合” 到附着位点(即着陆位点)上。在自然界中,这些酶识别微生物序列,因此 Hsu 对其进行工程化改造,使其可直接识别人体序列,无需预先插入着陆位点。
正如 2025 年《自然・生物技术》论文所述,Hsu 改造了重组酶蛋白、优化供体 DNA,并加入与 Cas9 的融合体,将重组酶拴在目标供体与靶 DNA 位点上。最终实现超过 50% 的插入效率与 97% 的特异性。

这些工程化大型丝氨酸重组酶可在多种细胞中工作(人胚胎干细胞、诱导多能干细胞、原代人 T 细胞),并可插入高达 12kb 的载荷。
Hsu 还在开发另一组无关的重组酶,他称之为桥接 RNA(bridge RNA)—— 因为这条向导 RNA 能同时识别两种不同 DNA 分子:供体与靶点。
而 CRISPR 的向导 RNA 只识别靶点 DNA,依赖内源机制寻找并将供体 DNA 插入切口。
他在两篇《自然》与一篇近期《科学》论文中描述了该系统。


据 Hsu 介绍,这是首个 RNA 向导可编程重组酶可实现通用 DNA 重排的系统:通过改变桥接 RNA 结合的 DNA 链,可实现 DNA 切除或翻转。借助该平台,Hsu 成功切除了导致弗里德赖希共济失调(Friedreich’s ataxia)的重复扩增序列,并切除与翻转人类基因组中巨大的 DNA 片段。
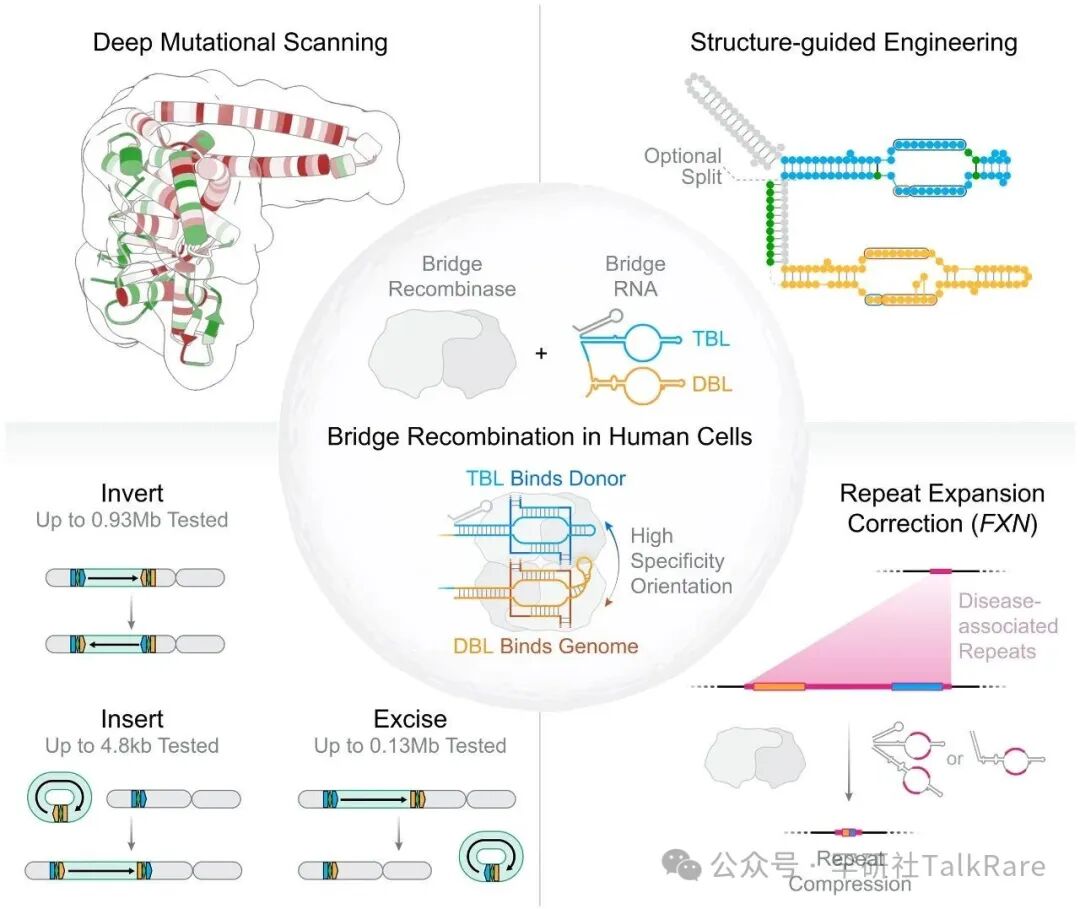
图:开发用于人类细胞中 Mb 规模基因组重排的桥接重组酶(引自:PMID: 40997214)
该系统仅含两个组分:重组酶(仅 300 多个氨基酸,约为 Cas9 的 1/4 大小);桥接 RNA。这意味着有多种方式可将这些组分高效递送至各类人体细胞。
Tessera 利用自然界的一类转座元件 ——逆转座子(又称 “跳跃基因”)构建了 “基因写入器”。这些可移动遗传元件通过 “剪切 - 粘贴” 机制自我复制并插入基因组。
公司利用这些元件设计出基因写入器多肽:可在特定位点结合 DNA、产生单链切口,并结合 RNA 模板启动逆转录。他们评估了超过 10 万种不同组合的基因写入器,筛选出具备特定活性的分子。
Tessera 首席执行官 Michael Severino 表示,其系统可纠正基因组任意位置的单核苷酸多态性,并以单碱基精度写入更长片段 —— 外显子长度乃至完整基因片段。
基因写入器以 mRNA + RNA 模板的双组分形式递送。
其初期项目针对:肝脏相关疾病:α1 - 抗胰蛋白酶缺乏症(AATD);造血干细胞相关疾病:镰状细胞病。
11 月,Tessera 宣布与再生元(Regeneron)合作,共同开发核心候选药物 TSRA-196,用于 AATD,以脂质纳米颗粒递送。
该合作基于 AATD 小鼠模型(PiZ 小鼠)与非人灵长类的临床前数据:小鼠中实现 95% 纠正,非人灵长类中,76% 的肝细胞实现 SERPINA1 位点纠正。
该基因的单点突变会产生毒性错误折叠蛋白,无法输送至肺部,导致肝脏炎症、纤维化以及肺部损伤。Severino 预计今年将进入临床。
宾夕法尼亚儿童医院的 Kiran Musunuru 与 Rebecca Ahrens-Nicklas 则采取了另一种精准基因编辑路线 —— 他们正是 KJ 婴儿疗法的设计者。
那次巨大成功需要 11 家机构协作,因此团队如今致力于开发一套可用于纠正多种突变驱动疾病的总方案(master protocol)。
在收到 “同行明确反馈:单一患者疗法无法产生真正能支持获批的科学证据” 后,他们正推动采用伞式临床试验的监管路径[参见:如何为罕见病患者打造个性化基因编辑平台?]。
他们提议将 7 种尿素循环障碍纳入同一试验 —— 每种疾病均破坏同一生化通路中不同蛋白,但可使用相同临床终点。
Musunuru 说:“在总方案中,唯一变化的只是向导 RNA(gRNA),根据变异所在的特定基因区域调整。递送载体与编辑 mRNA 保持一致。”
随着可纠正一组突变或一类疾病的疗法即将进入人体试验,监管机构需要重新思考以适应这类全新模式。
美国 FDA 两位高级官员近期提出的“合理机制路径”已接纳这类可能性。英国药品与医疗保健产品监管局(MHRA)也表示希望加速罕见病疗法的获批时间。美国政府在卫生高级研究计划局(ARPA-H)设立新项目:罕见病 AI/ML 精准整合诊断。
今年 1 月,加州再生医学研究所启动 1 亿美元项目 RAPID(通过平台创新与递送加速罕见病治疗),以加速罕见病基因疗法研发。
这个不依赖特定突变的治疗新时代,有望克服罕见病给患者与生物制药行业带来的巨大困境。
Hsu 对此乐观:“我们知道很多需要对基因组做出的改变,但一直无法实现 —— 而借助这些新系统,我们正在解锁全新能力,开始攻克其中一些极具挑战的难题。”

译者注:作者 Laura DeFrancesco 为资深科学记者。图片、文章版权均属于原作者所有,如有侵权,请及时告知,我们会在24小时内删除相关信息。欢迎转载,转载请注明出处。


















