在罕见神经遗传病的诊疗中,「查不出病因」是无数患者和家庭最大的困境。
肌张力障碍,是一类以持续性或间歇性肌肉收缩导致异常运动和/或姿势为核心特征的致残性运动障碍病,超半数患者在21岁前发病,且绝大多数具有明确的单基因遗传倾向。但长期以来,即便用上了临床常规的外显子测序(ES)、短读长全基因组测序(srGS),仍有超过70%的患者始终无法获得明确的基因诊断,陷入「临床高度怀疑遗传病,却找不到致病基因」的死胡同。

近期,国际运动障碍领域顶刊Movement Disorders发表了一项来自德国慕尼黑工业大学、亥姆霍兹慕尼黑中心等机构的重磅研究,首次系统验证了牛津纳米孔技术(ONT)长读长全基因组测序(lrGS) 在疑难肌张力障碍诊断中的突破性价值:14例经传统短读长测序无法确诊的疑难病例,13例通过长读长测序获得了明确的分子诊断,1例成功排除了假阳性致病结果,一举解决了传统测序几乎所有的核心技术短板。
传统测序的瓶颈:为什么78%的肌张力障碍患者查不出病因?
这项研究基于一个超大规模的肌张力障碍研究队列,纳入了1825例患者,数据触目惊心:
经临床金标准外显子测序检测后,仅21.7%的患者获得明确诊断,78.3%的患者无明确致病结果;
针对305例外显子测序阴性的患者,进一步加做短读长全基因组测序,诊断率也仅提升了12.1%,仍有87.9%的患者无法确诊。
为什么看似先进的二代测序,在肌张力障碍面前频频失灵?
核心原因在于,短读长测序的技术原理存在无法突破的固有局限:它只能读取长度150bp左右的DNA片段,就像用拼图碎片还原整幅画作,面对基因组里的复杂区域,天然存在看不清、测不准、辨不明的问题:
无法获取长范围单倍型信息,无法确定两个杂合变异的顺反式构型,复合杂合变异无法确诊;
对结构变异(SV)、复杂结构重排的分辨率极低,单外显子缺失、插入、倒位等变异频繁出现假阳性/假阴性;
对串联重复扩增(TRE)这类动态突变,只能估算重复数,无法精准定量,更无法识别嵌合体;
面对存在高度同源假基因的区域,短序列无法区分真基因与假基因,频繁出现错配和误判;
对高GC含量的基因组区域覆盖度极差,大量致病变异被直接漏检;
无法同时获取DNA序列与甲基化修饰信息,意义不明确的变异(VUS)无法完成致病性重分类。
而这些短板,恰恰是肌张力障碍致病基因变异的「重灾区」。
长读长测序的六大核心优势,一站式解决诊断难题
这项研究中,研究团队针对14例疑难病例,通过ONT长读长测序完成了「从无解到确诊」的突破,也淋漓尽致地展现了长读长测序在遗传病诊断中的核心价值。
优势一:无需家系样本,一键完成变异定相,锁定复合杂合致病
常染色体隐性遗传病的确诊核心,是确认两个杂合变异分别来自父母、位于两条染色体上(反式构型,即复合杂合)。但传统短读长测序无法跨越变异位点,必须通过父母样本测序完成定相,而很多患者因亲属离世、无家属联系方式等原因,无法完成家系检测,诊断就此停滞。
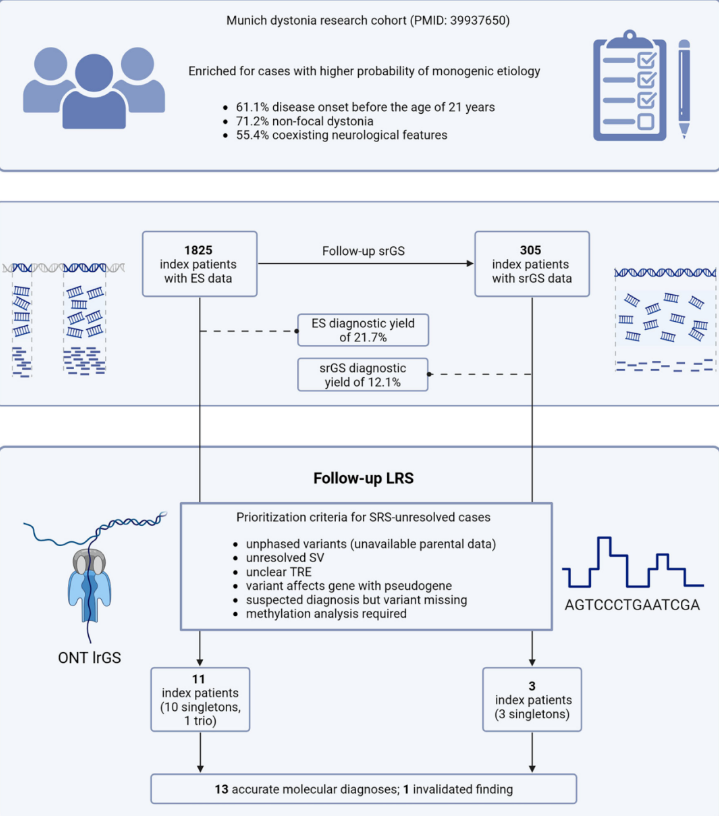
本研究中,4例患者均因无家系样本,无法确认PNPT1、SZT2、TH、HIBCH基因上的变异构型。长读长测序通过超长reads直接跨越两个变异位点,无需任何家系样本,一次性确认了4例患者的变异均为反式复合杂合,分别确诊了线粒体病、发育性癫痫性脑病、Segawa综合征、3-羟基异丁酰辅酶A水解酶缺乏症。
其中2岁的男性患儿,表现为肌张力障碍、肌阵挛、震颤,临床高度怀疑多巴反应性肌张力障碍,传统测序仅发现TH基因两个杂合变异,无法定相确诊。长读长测序确认复合杂合后,患儿最终确诊Segawa综合征,这类疾病可通过左旋多巴治疗获得显著改善,精准诊断直接为患儿打开了治疗的大门。
优势二:结构变异的「高清显微镜」,精准解析复杂重排,发现全新致病机制
结构变异(大片段缺失、重复、倒位、插入)是肌张力障碍的重要致病原因,但传统测序对这类变异的解析能力几乎为零,要么无法识别,要么无法明确断裂位点和变异构型。
研究中,3例患者通过长读长测序完成了结构变异的终极解析:
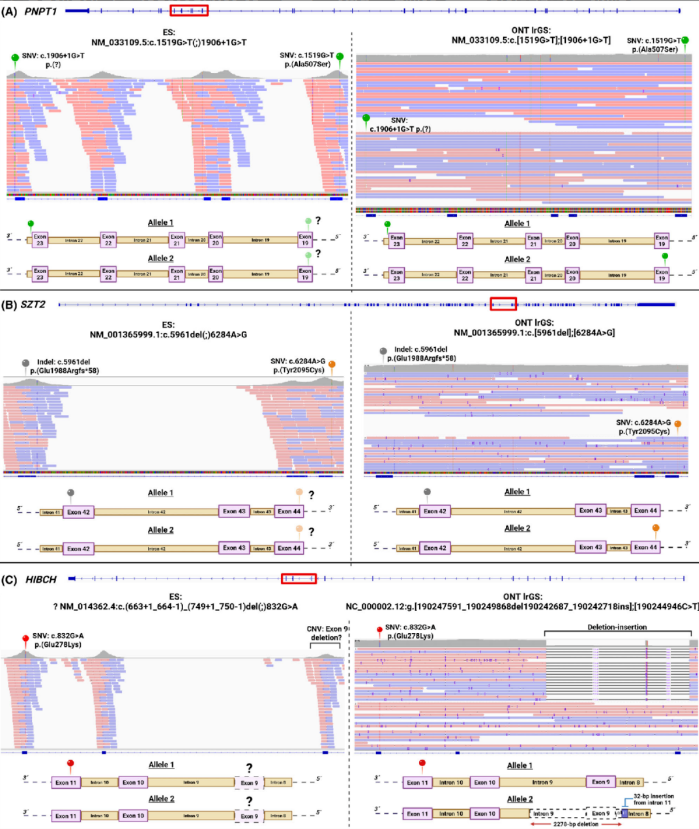
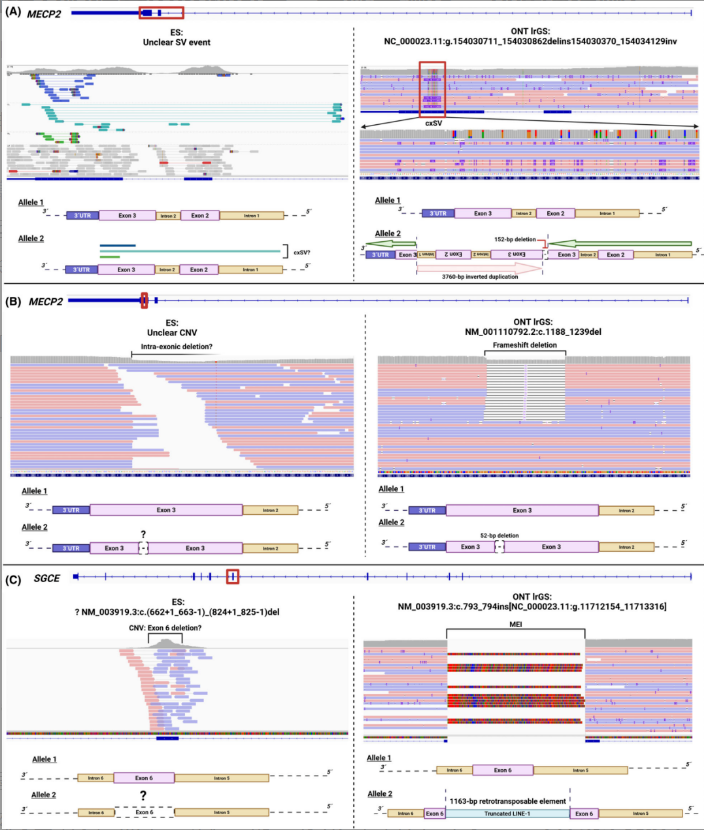
临床高度怀疑Rett综合征的患者,传统测序仅提示MECP2基因可能存在复杂结构变异,但无法明确细节。长读长测序精准解析了变异全貌:3号外显子152bp缺失,同时伴随包含2、3号外显子的3760bp倒位重复,还确认了该变异为新发变异,直接确诊Rett综合征;
肌阵挛-肌张力障碍患者,传统测序提示SGCE基因6号外显子杂合缺失,但这类单外显子缺失在传统测序中假阳性率极高。长读长测序最终证实,该区域并非缺失,而是插入了一段1163bp的LINE-1转座子,这是SGCE基因全新的致病机制,直接为患者明确了诊断。
优势三:串联重复扩增的「精准标尺」,破解动态突变与嵌合体难题
超过30种神经系统遗传病由串联重复扩增(动态突变)导致,包括脊髓小脑共济失调、脆性X综合征等,这类变异也是肌张力障碍的重要病因。传统测序只能通过算法估算重复数,对接近致病阈值、超长重复、嵌合型重复的检测完全失效。
本研究中3例患者的串联重复变异,均通过长读长测序实现了精准诊断:
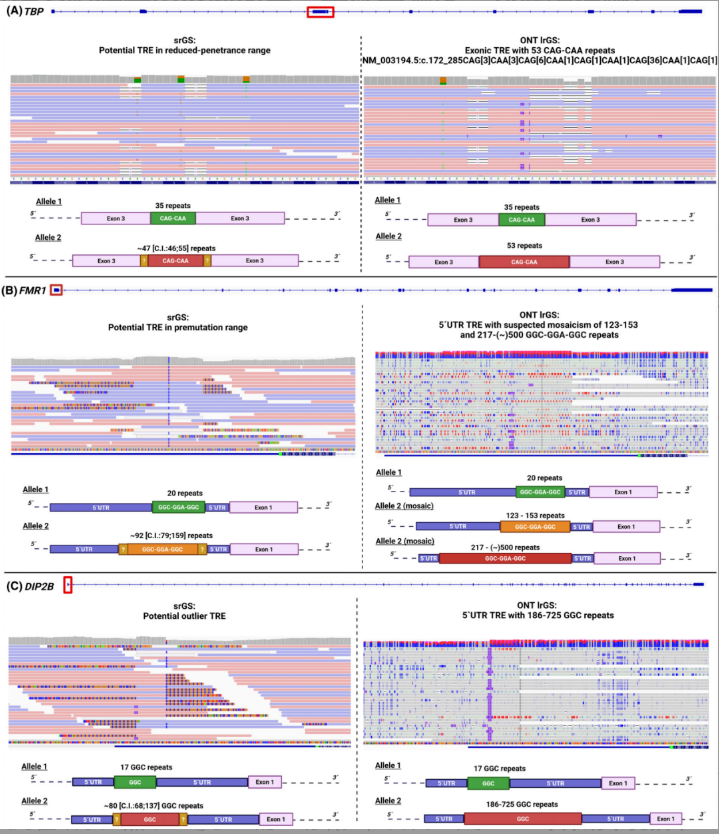
成人起病的肌张力障碍-帕金森综合征患者,传统测序估算TBP基因CAG重复数为47次,处于低外显率区间,无法确诊。长读长测序精准测出重复数为53次,明确超过致病阈值(>48次),确诊脊髓小脑共济失调17型;
局灶性孤立性肌张力障碍患者,传统测序估算FMR1基因重复数为92次,仅提示前突变。长读长测序发现该患者存在嵌合变异,既有120-150次的前突变,也有200-500次的全突变,同步甲基化分析确认了高甲基化状态,最终明确了表型与基因型的关联。
优势四:破解假基因陷阱,杜绝假阳性/假阴性误诊
很多致病基因存在高度同源的假基因,短读长测序的短序列无法区分真基因与假基因,极易将假基因的序列错误比对到真基因上,导致假阳性的纯合变异诊断,给患者带来误诊风险。
研究中1例患者,传统测序提示PI4KA基因存在纯合错义变异,与患者的肌张力障碍、痉挛性瘫痪、脑白质异常表型完全匹配,几乎就要确诊。但PI4KA基因存在两个高度同源的假基因,长读长测序最终证实,该变异实际为杂合状态,传统测序因假基因的序列错配,误判为纯合变异,直接排除了这一假阳性诊断,避免了后续的错误诊疗。
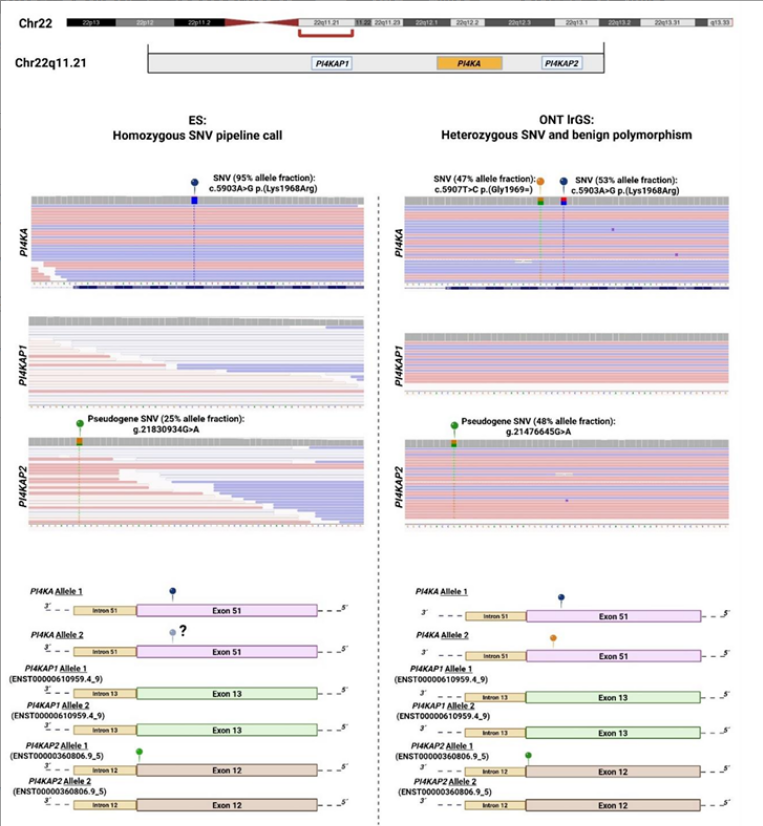
优势五:覆盖传统测序「盲区」,找到漏检的致病变异
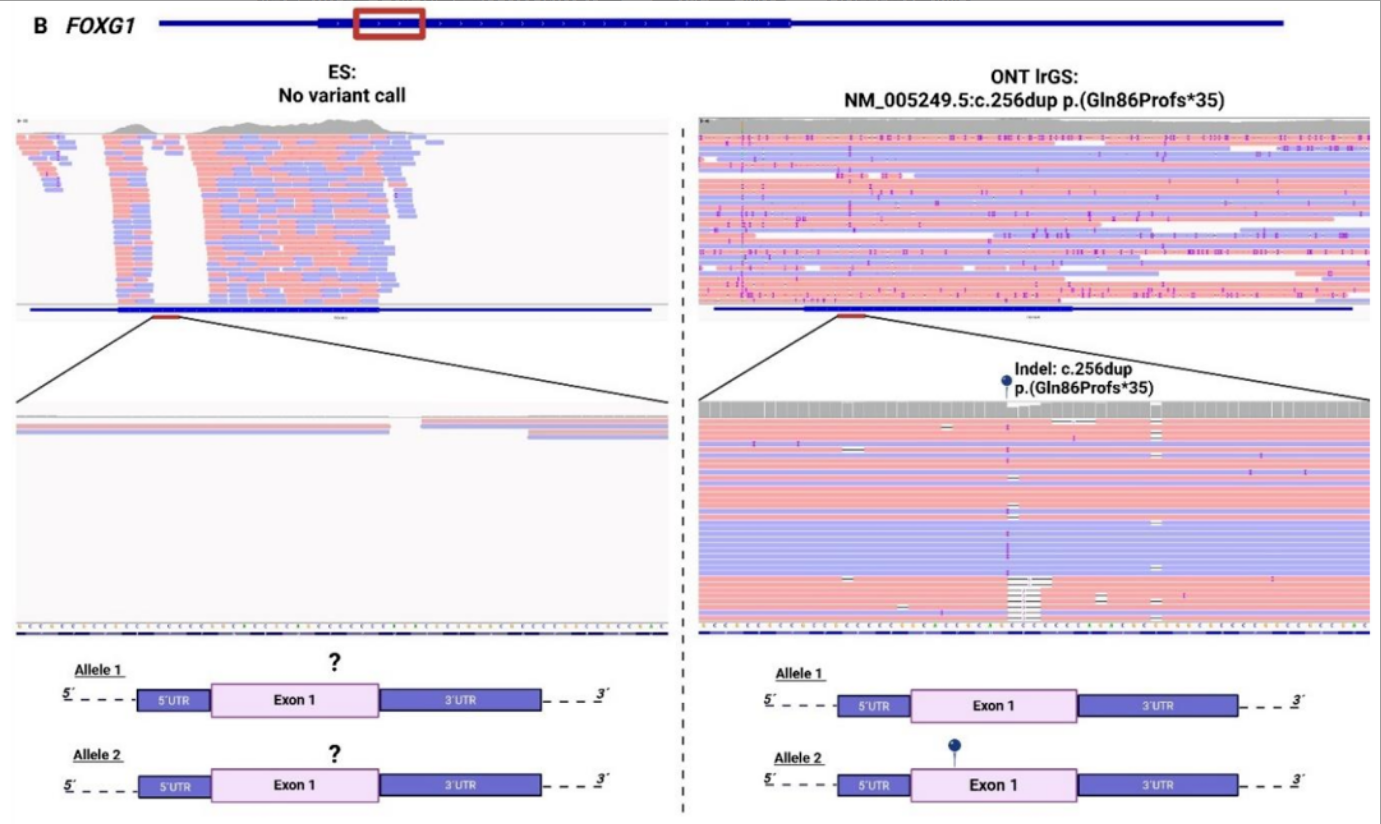
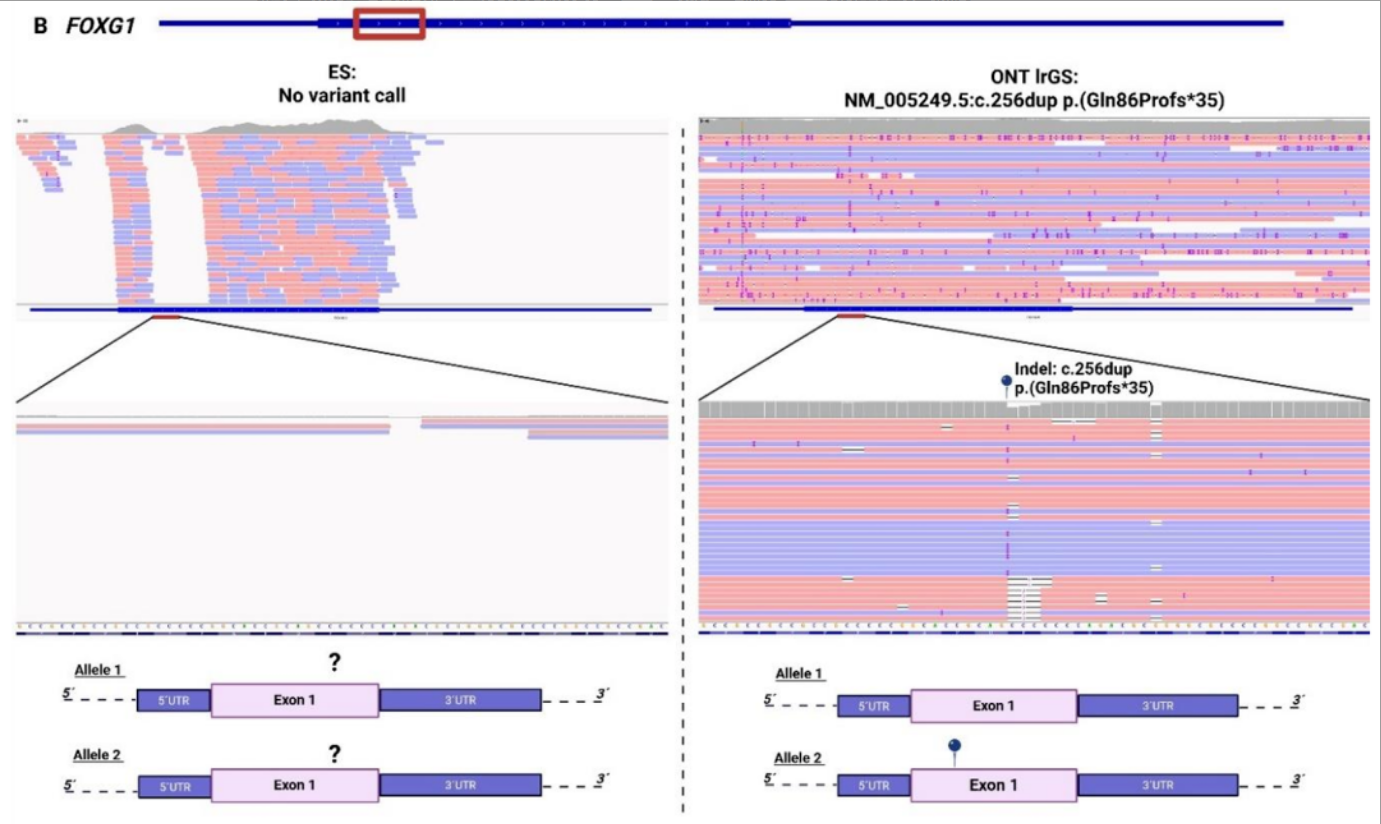
传统外显子测序对高GC含量的基因组区域,往往存在覆盖度极低甚至无覆盖的问题,这些区域的致病变异会被直接漏检,成为诊断的「盲区」。
1例临床高度怀疑先天性Rett综合征的患儿,传统外显子测序反复检测均无阳性发现。长读长测序最终在FOXG1基因的高GC富集区(传统测序完全无覆盖),找到了致病性的插入缺失变异,为患儿明确了先天性Rett综合征的诊断,结束了漫长的诊断寻因之路。
优势六:一次测序,同步获取基因组+甲基化组数据,一站式解决VUS重分类
临床测序中,约30%-50%的结果会检出意义不明确的变异(VUS),无法确定其致病性,也无法用于确诊。而基因特异性的甲基化特征(表观签名),是判断变异致病性的核心依据之一。传统测序若要完成甲基化分析,需要额外开展甲基化芯片或重亚硫酸盐测序,成本高、流程复杂。
而ONT长读长测序,仅需一次测序,就能同时获取DNA序列信息与单碱基分辨率的甲基化修饰信息,无需额外实验。研究中1例患者,传统测序检出KMT2B基因的一个框内插入缺失VUS,无法确诊。长读长测序不仅验证了变异,还同步完成了80个CpG位点的甲基化分析,发现患者的甲基化模式与KMT2B相关肌张力障碍的特征性表观签名高度一致,直接将该VUS重分类为致病性变异,为患者明确了诊断。
长读长测序:遗传病诊断的下一个黄金时代
这项研究,是首个系统验证ONT长读长测序在肌张力障碍诊断中临床价值的重磅工作,它用实打实的临床数据证明:长读长测序并非传统测序的「锦上添花」,而是解决疑难遗传病诊断困局的「关键钥匙」。
14例传统测序无法解决的疑难病例,13例明确诊断,1例排除假阳性,这样的诊断效能,彻底打破了传统测序的技术天花板。它能一站式解决传统测序的几乎所有核心短板:变异定相、结构变异解析、串联重复精准定量、假基因区分、盲区覆盖、甲基化联合分析,真正实现了「一次测序,全维度解析」。
对于临床医生而言,面对传统测序阴性的疑难肌张力障碍、神经遗传病患者,长读长测序理应成为重要的二线检测手段,甚至在高度怀疑动态突变、结构变异相关疾病时,可作为一线检测方案。
对于无数罕见病患者和家庭而言,长读长测序带来的,不仅是一个明确的诊断,更是结束诊断奥德赛的希望、精准治疗的前提、遗传咨询与优生优育的依据,更是对「为什么是我」这个终极问题的答案。
随着技术的不断成熟和成本的持续下降,长读长测序正在从科研走向临床,从「小众技术」变为「常规工具」。我们有理由相信,长读长测序的普及,终将让更多罕见病患者走出「查不出病因」的困境,迎来精准诊疗的曙光。


















